
Evaluation of [11C]ER176 for imaging translocator protein (TSPO)
- Introductory statement of purpose and general plan
- Study protocol: Assessment of [11C]ER176 to image translocator protein in brain and whole body of healthy subjects
- Brain and lung imaging with [11C]ER176 (Phase 1)
- Whole-body imaging with [11C]ER176 for dosimetry (Phases 0 and 2).
- Chemistry, manufacture, and control
- Pharmacology and toxicology
- Animal experimentation
- Human experience
- Environmental assessment
- Case report form
- References
- APPENDICES
Introductory statement of purpose and general plan
-
Overview
Translocator protein 18 kDa (TSPO) is highly expressed in activated microglia and reactive astro-cytes in brain, and may thus be a useful biomarker of neuroinflammation. We developed [11C]PBR28 as a positron emission tomography (PET) radioligand to bind to TSPO and measure its density, but [11C]PBR28 is sensitive to TSPO affinity states both in vitro and in vivo. Here, we seek to assess the new TSPO radioligand [11C]ER176 for TSPO binding in brain and whole-body of healthy subjects, and measure its sensitivity to TSPO affinity states in vivo.
Although [11C]PBR28 has a high in vivo specific signal, it is very sensitive to the high and low affinity states of TSPO, which are caused by a single nucleotide polymorphism (SNP) in the fourth exon of the TSPO gene. This co-dominant mutation yields three genetic groups: HH, HL, and LL, where H is the high-affinity form and L is the low affinity form. TSPO binding by [11C]PBR28 is categorized into high affinity binders (HABs), medium affinity binders (MABs), and low-affinity binders (LABs). The frequency of the L allele is ~30%; thus, the frequency of the LL homozygote is ~9%. The affinity of PBR28 to H and L forms differs about 50-fold; thus, no measureable signal in brain from [11C]PBR28 is detectable in LL carriers. In contrast, the af-finity of ER176 differs by only 1.2 fold, so LL carriers should have measureable brain uptake. [11C]ER176 is O-methyl-11C](R)-N-sec-butyl-4-(2-chlorophenyl)-N-methylquinazoline-2-carboxamide. Its structure is shown in Figure 1.
Figure 1. Structure of [11C]ER176Using [11C]ER176, we will study up to 36 healthy adult female and male volunteers (18 years and older) for brain imaging; approximately 12 volunteers will be drawn from each of the three TSPO genotypes. In about half of the subjects from each genotype group, lungs will be scanned in the same session to characterize binding sensitivity to TSPO affinity states. In addition, we will study a separate group of up to 11 healthy adult female and male volunteers (18 years and older) for whole body dosimetry analysis. We request permission to administer a single injection of [11C]ER176. The injected activity will be ≤ 20 mCi, with an associated mass of ≤ 5 μg per sub-ject per injection. For a 70 kg subject, this would correspond to a maximum mass dose of 0.07 μg/kg per injection.
-
Statement of Purpose
The purpose of this study is to assess the potential of [11C]ER176 to image TSPO in brain, characterize its binding sensitivity in the lungs of healthy subjects from all three genetic groups, and do whole-body imaging for biodistribution and estimation of radiation dosimetry in humans. To achieve this purpose we will: 1) perform an initial whole-body scan after [11C]ER176 injection in a single healthy volunteer to confirm widespread distribution of radioactivity to different body organs (Phase 0); 2) perform kinetic brain scans in healthy volunteers from three different geno-types, with about half of these volunteers undergoing lung scans in the same session (Phase 1); and 3) perform whole-body imaging in a separate group of healthy volunteers (Phase 2).
Study protocol: Assessment of [11C]ER176 to image translocator protein in brain and whole body of healthy subjects
The complete protocol is located in Appendix B. Brief summaries are included below.
Brain and lung imaging with [11C]ER176 (Phase 1)
Objective: To characterize brain uptake and distribution of [11C]ER176 and measure in vivo binding sensitivity of TSPO by lung imaging.
Study population: Healthy volunteers (n = up to 36).
Design: Healthy subjects from three TSPO genotypes will be recruited via a screening protocol that includes a medical and psychiatric history, physical exam, and laboratory testing. Results of the screening will be used to determine whether subjects qualify as healthy volunteers. Each eli-gible subject will have a brain magnetic resonance imaging (MRI) scan (for anatomic localization of structures) and one PET or PET/CT scan. Prior to the PET scan, an anesthesiologist will insert a radial arterial catheter for subsequent blood sampling. The radioligand [11C]ER176 will be in-jected intravenously up to 20 mCi, and serial brain images and arterial plasma samples will be ac-quired for about 2 – 2.5 hours to determine if tracer uptake can be quantified relative to concur-rent plasma concentration of the radiotracer. Because lungs may provide better sensitivity of TSPO binding, brain and lungs will both be imaged in the same scan session for about half of the subjects in each genotype group
Outcome measures: To assess absolute quantitation of TSPO in brain with [11C]ER176, we will primarily use two outcome measures: the identifiability and time stability of distribution volume (VT) calculated with compartmental modeling. As a secondary outcome measure, we will examine the effect of polymorphisms on [11C]ER176 binding in lungs because lungs have a much higher density of TSPO and may be more effective in assessing whether ER176 is sensitive to the SNP.
Whole-body imaging with [11C]ER176 for dosimetry (Phases 0 and 2).
Study population: Healthy volunteers (n = up to 11).
Design: Healthy subjects will be screened for inclusion in the study, which will include a medical and psychiatric history, physical exam, and laboratory testing. Results of the screening will be used to determine whether subjects qualify as healthy volunteers. Each eligible subject will un-dergo whole body PET imaging in PET or PET/CT for about two to three hours after intravenous injection of up to 20 mCi of [11C]ER176, in order to estimate radiation exposure to organs of the body. These dosimetry studies are required to confirm the radiation safety of the compound.
Outcome measures: Organ time-activity curves will be obtained from the dynamic PET images in order to calculate organ residence times. The residence times will be used in OLINDA/EXM (http://www.doseinfo-radar.com/OLINDA.html) to obtain radiation-absorbed dose estimates.
Chemistry, manufacture, and control
The CMC Section is included in Appendix E.
Pharmacology and toxicology
Pharmacology
Because TSPO is highly expressed in microglia and reactive astrocytes, it has been widely used over the last three decades as an in vivo biomarker to detect neuroinflammation using PET (Stephenson 1995). In recent years, a large number of clinical studies have been performed using new PET ligands, including [11C]PBR28, which was developed by our group. Although these new PET ligands have higher levels of specific binding than the classic ligand [11C]PK 11195 (Chauveau 2008), they are confounded by the fact that the binding affinity of most new ligands is affected by TSPO polymorphisms (Owen 2012). As noted above, SNPs determine whether human subjects are high- (HAB), mixed- (MAB), or low-affinity (LAB) binders. PET results may thus be determined by genotype in addition to pathological changes in TSPO density (Kreisl 2012).
To cope with the confounding factor of polymorphism, we developed a new ligand, [11C]ER176, with similarly high affinity for all three genotypes. Ki values of ER176 and [H-3]PK 11195 measured in human leukocytes were 1.4 and 1.6 nM for HAB and LAB, respectively (Fig. 2). ER176 also has lipophilicity of clog D = 3.80, which is appropriate for brain imaging.
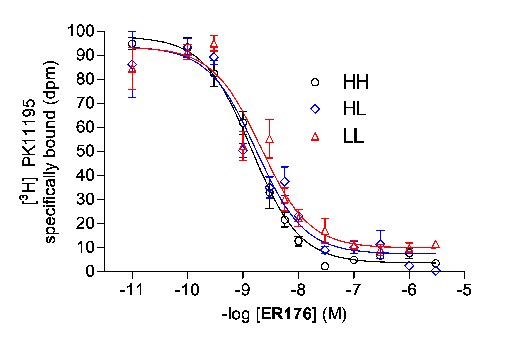
Fig. 2. Displacement of [H-3]PK 11195 with ER176 in HH, HL, and LL leukocytesToxicology
SRI International conducted an extended acute toxicity study in rats. The executive summary is provided below, and the complete report is located in Appendix F. Please note that the toxicity study was designed around the assumption that the maximal mass dose would be 10 µg in a human subject. After receiving the report, and to be extra cautious, we reduced the max-imal mass dose to 5 µg. Thus, the report and this IND differ two-fold when referring to the pro-jected human dose. For example, in the report, the actual dose of 22.0 µg/kg in a rat is referred to as 25X human equivalent dose (HED), whereas this IND refers to it as 50X HED.
Executive Summary
The objective of this study was to determine potential toxic effects and to identify potential tar-get organs of toxicity, if possible, for the toxicity endpoints examined following a single intrave-nous (iv) bolus administration of ER176 given to Sprague Dawley rats. Additionally, the maxi-mum tolerated dose (MTD) and no observed adverse effect level (NOAEL) of ER176 in rats may be established. Information from this study may be used to determine the suitability of the proposed human dose.
Two groups of 10 male and 10 female rats were administered a single dose of either the vehicle control (Group 1) or ER176 at 88.1 µg/kg (528.6 µg/m2; 100 times the human dose; Group 2) on Day 1 by iv bolus injection into the tail vein. The vehicle was 10% ethanol and 90% saline. In Groups 1 and 2 five males and five females in each group were sacrificed on Day 3, while the remaining animals were sacrificed on Day 15. In-life evaluations of mortality, morbidity and clin-ical observations were conducted daily. Body weights were collected on Days 1, 3 and 15. Food consumption was evaluated twice weekly. Clinical pathology samples were collected on the day the animals were euthanized. All animals survived to their scheduled sacrifices. There were no test article–related effects seen in the animals treated with 88.1 µg/kg ER176 on body weights, food consumption, clinical pathology, gross necropsy, organ weights or histopathology, com-pared with the controls. However, slight ataxia was observed immediately post dose administra-tion in 10 of 10 males and 6 of 10 females in the ER176 treated group. The effect was slight and transient, and the animals recovered quickly (within 2 hr post dose administration). All animals in the control group were normal. Due to this clinical finding and in an attempt to establish the NOAEL, additional animals were evaluated for clinical observations after consultation with the Sponsor.
Three groups of 10 male and 10 female rats were administered ER176, by iv tail vein injection, at 22.0 µg/kg (132.5 µg/m2; 25 times the human dose; Group 3), 44.1 µg/kg (264.3 µg/m2; 50 times the human dose; Group 4), or 88.1 µg/kg (528.6 µg/m2; 100 times the human dose; Group 5). In-life evaluations of mortality, morbidity and clinical observations were conducted daily for 3 days, and animals were sacrificed on Day 3. No test article–related effects were observed in the animals treated with 22.0 or 88.1 µg/kg of ER176 (Groups 3 and 5). However, a slight and tran-sient ataxia was observed in one male rat immediately following dosing in the 44.1 µg/kg dose group (Group 4). It is not clear why animals in the high dose groups (100 times the human dose; 88.1 µg/kg) responded differently to ER176 in Groups 2 and 5 in clinical observations immedi-ately post dose administration. It may be due to animal-to-animal variations in different ship-ments.
In conclusion, a single dose of ER176 (22.0 µg/kg, 132.5 µg/m2; 25 times the projected human dose) administered intravenously was well tolerated in rats and produced no significant treat-ment–related effects. Animals in the 44.1 and 88.1 µg/kg treatment groups also tolerated the sin-gle dose administration, except a slight and transient ataxia was observed. The NOAEL is con-sidered to be 22.0 µg/kg (132.5 µg/m2; 25 times the projected human dose). The MTD is consid-ered to be greater than 88.1 µg/kg (528.6 µg/m2; 100 times the projected human dose).
Requested Mass Dose
We request permission to inject a maximal mass dose of 5 µg, which is two-fold less than the 10 µg that was considered the maximal dose for toxicity in the study described above. Below, we explain the rationale for this request and its expected safety.
Dose Conversions: The NOAEL for ER176 in rats is 22 µg/kg i.v. The following Table converts this and other tested doses to the HED, based on a projected human weight of 70 kg and species conversion based on surface area. For this species conversion, HED = 0.162 * dose in rats
Rat Dose (µg/kg) HED (µg/kg) Dose for 70 kg human (µg) Dose for human / dose for rata 88.1 14.3 999 200 X 44.1 7.1 500 100 X 22.0 3.6 250 50 X a Considers a mass dose of 5 µg in humans
Background Information: The guidance for the exploratory IND (January 2006) appears to allow an NOAEL of less than 100 X HED because it provides that value only as an example. See highlighted text below.
“Microdose studies are designed to evaluate pharmacokinetics or imaging of specif-ic targets and are designed not to induce pharmacologic effects. Because of this, the risk to human subjects is very limited, and information adequate to support the initi-ation of such limited human studies can be derived from limited nonclinical safety studies. A microdose is defined as less than 1/100th of the dose of a test substance calculated (based on animal data) to yield a pharmacologic effect of the test sub-stance with a maximum dose of <100 micrograms (for imaging agents, the latter cri-terion applies). To establish a margin of safety, the sponsor should demonstrate that a large multiple (e.g., 100X) of the proposed human dose does not induce adverse effects in the ex-perimental animals.”
The toxicity study in rats demonstrated a NOAEL of 50X the maximal human dose of 5 µg.
Rationale for Safety
- NOAEL. From a strict perspective, the NOAEL is 25X HED, but this was not a robust and reproducible finding. The initial study noted ataxia in most animals injected at 100X HED, but the finding was not reproducible. We repeated the study at 100X HED and added 50X HED and 25X HED. In this case, none of the animals at 100X HED and only one of the animals at 50X HED showed ataxia. Please note that we used the same batch and formulation of the test compound. Thus, this behavioral finding was marginal and presumably dependent on the somewhat variable condition of the test animals.
- Transient Effect. The initial study at 100X HED noted ataxia immediately after a bolus IV injection of less than one minute. The next clinical observation was only two to four hours later, and by then all animals were normal. For the repeat study at 100X, 50X, and 25X, clinical observations were performed immediately post-dose and approximately 15, 30, 60, 90, and 120 minutes post-dose to more carefully follow the time course of ataxia. While no ataxia was noted in the repeat 100X group, slight ataxia was observed immedi-ately post-dose in one of 10 male rats in the 50X HED dose group. This animal returned to normal by the second clinical observation time point (15 minutes post-dose) . The atax-ia was transient and occurred immediately after administration. The transient effect sug-gests that the ataxia was related to high plasma concentrations immediately following in-jection. The conversion for HED tries to account for total exposure of the drug, which is related to metabolism and is better correlated with body surface area than with body weight. However, the initial plasma concentrations following IV injection are primarily affected by distribution (a function of total volume available to the drug), which corre-lates in the opposite way – namely, better with body weight than with surface area. If we calculate the NOAEL of 22 µg/kg in rat based on body mass, the HED for a 70 kg sub-ject would be 1540 µg. Please note that our proposed dose of 5 µg is two-fold lower than this revised NOAEL.
- Decreased Infusion Rate. Because the effects of ataxia are likely related to high plasma concentrations immediately after IV injection, we will decrease the infusion rate in human subjects. For all of our other INDs for radiopharmaceuticals, the maximal mass dose is 10 µg, whereas we now request only 5 µg. In addition, we will also increase the duration of injection from a typical period of one minute to three minutes. We will inject the radiolig-ands via a Harvard pump, which maintains a constant rate over the entire injection period. Thus, the combination of injecting half the maximal dose over a three-time longer period would likely decrease peak plasma concentrations by about six-fold.
- Extra Monitoring of Subjects. We will, of course, inform subjects of these behavioral findings in rats as part of the consent process. We will also mention that the effects may be transient and occur within the first few minutes of the study and ask them to be par-ticularly vigilant of any subjective effects during the early portion of the study. At the end of the study, we will ask the person to gradually stand up. We will be there to assist as needed and ask if they have any difficulty standing or walking. We will release sub-jects only after we— and they—feel entirely normal and are walking normally.
Animal experimentation
PET imaging using [11C]ER176 in monkeys
We performed a pair of baseline and pre-blocked PET scans of [11C]ER176 in two rhesus monkeys (a total of two baseline and two pre-blocked scans) and confirmed that [11C]ER176 has high levels of specific binding. In both monkeys, binding blockade was measured using brain ac-tivity. In addition, in one monkey, a more complete measurement of binding was performed by measuring total VT using metabolite-corrected arterial input function, i.e., [11C]ER176 levels in arterial plasma. For the pre-blocked scans, PK 11195 (5 mg/kg) was used as the blocking agent. In the baseline scans, the mass dose of ER176 was 0.111 µg/kg on average. The scans were per-formed under isoflurane anesthesia; blood pressure, heart rate, and ECG were monitored. These mass doses caused no changes in vital signs.
Based on brain activity at late time points (60 - 120 minutes), 60% of brain activity in baseline scans was from specific binding (Fig. 3). However, these measurements did not take into account changes in blood data. VT measured with arterial input function and Logan plot was 11 – 17 mL/cm3 in baseline and 1.6 – 2.5 mL/cm3 in pre-blocked scans. The complete measurements suggest that ~85% of activity of the baseline scan was specific binding.

Figure 3. Mean time activity curves in monkey brain at baseline (blue) and pre-blocked (red) with PK 11195 scansAbsorption, Distribution, and Metabolism
In the baseline and pre-blocked scans of one monkey described in section A, we measured [11C]ER176 levels in arterial plasma (Fig. 4). The pre-blocked scan showed about five times high-er [11C]ER176 levels in arterial plasma. TSPO exists in several peripheral organs such as lungs, heart, and kidneys. We performed whole body imaging and arterial blood sampling at the same time with another ligand ([11C]PBR28), and confirmed that [11C]PBR28 increases in arterial plasma were caused by binding blockade to TSPO in peripheral organs (Imaizumi 2008).
Therefore, increased [11C]ER176 levels in arterial plasma in the pre-blocked scan is consistent with the presence of specific binding to TSPO in peripheral organs.
The arterial data from the two monkey scans showed fast clearance of [11C]ER176, the con-centration of which decreased to a half of the peak within three minutes post-injection and to < 10% in 30 minutes. Clearance was 526 and 195 mL/min for the baseline and blocked scan, and the terminal half life by tri-exponential fitting was 24 and 49 minutes, respectively.
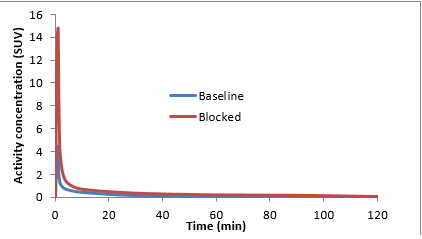
Figure 4. [11C]ER176 levels in arterial plasma in baseline (blue) and pre-blocked (red) scans.Human experience
Safety
ER176 has never been administered to humans; its safety is thus unknown.
Dosimetry
Radiation dosimetry of [11C]ER176 is not available for humans. However, based on argu-ments made in a previous publication (Zanotti-Fregonara 2012), we think that animal dosimetry results are unnecessary and potentially misleading for 11C-radioligands.
Among 21 different C-11-labeled PET ligands, the highest effective dose (16 μSv/MBq) was found for [11C]WAY-100635, and the average effective dose was ~ 5 μSv/MBq. Although the International Commission on Radiological Protection (ICRP) has not proposed any radiation dose limits for volunteer subjects in human radiation experiments, the Institutional Radiation Safety Committees usually do establish upper limits. For instance, at the National Institutes of Health (NIH), the upper limit of the effective dose for subjects participating in research radiation studies is 50 mSv (http://www.ors.od.nih.gov/sr/drs/Documents/informed_consent.pdf). Even with ligands such as [11C]WAY-100635, the maximum injectable activity to reach an upper limit of 50 mSv effective dose would be 3125 MBq (84 mCi). However, the limiting organ for [11C]WAY-100635 is the bladder, with an estimated dose of 0.167 mGy/MBq in men and 0.220 mGy/MBq in women (Parsey 2005). Even then, an activity of up to 300 MBq in men and 227 MBq in women can be administered in a single injection before reaching 50 mSv, an organ limit established by the US Food and Drug Administration for radioligands studied under a Radioac-tive Drug Research Committee (21CFR361.1).
In view of the above dosimetry considerations for a C-11-labeled ligand, we propose to initi-ate human studies without animal dosimetry. Instead, we plan to scan the first human subject with a whole-body scan to confirm that radiation exposure is similar to that of published studies with 11C-radioligands – namely, an effective dose within the range of 3 to 16 µSv/MBq (Zanotti-Fregonara 2012).
Environmental assessment
A categorical exclusion is requested regarding the environmental assessment. Activity excret-ed by the subjects will be disposed through the sewage system at such low amounts that there will be no detrimental effects to the environment.
All potentially contaminated laboratory materials will be held at least 10 half lives for decay. In addition, these materials are surveyed to ensure background levels of activity prior to disposal through regular waste systems.
Case report form
The case report form is in Appendix D.
References
Chauveau, F., et al. (2008). "Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers." Eur J Nucl Med Mol Imaging 35(12): 2304-2319.
Imaizumi, M., et al. (2008). "Brain and whole-body imaging in nonhuman primates of [11C]PBR28, a promising PET radioligand for peripheral benzodiazepine receptors." Neuroimage 39: 1289-1298.
Kreisl, W. C., et al. (2012). "A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation." Journal of Cerebral Blood Flow and Metabolism.
Owen, D. R., et al. (2012). "An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28." J Cereb Blood Flow Metab 32(1): 1-5.
Parsey, R. V., et al. (2005). "Biodistribution and radiation dosimetry of 11C-WAY100,635 in humans." J Nucl Med 46(4): 614-619.
Stephenson, D. T., et al. (1995). "Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat." J Neurosci 15(7 Pt 2): 5263-5274.
Zanotti-Fregonara, P., et al. (2012). "Suggested pathway to assess radiation safety of (11)C-labeled PET tracers for first-in-human studies." Eur J Nucl Med Mol Imaging 39(3): 544-547.
APPENDICES
- CV of investigator
- Study protocol
- IRB protocol approval
The CNS (Combined Neurosciences) IRB is currently reviewing the protocol. We will not begin the study until it is approved by the IRB, and until the FDA has issued a “may proceed" letter for the IND.
-
Case report forms
Six pages case report form
- Chemistry, Manufacturing, and Controls
-
Toxicology data in rats
Only the first six pages of the report are provided here in hard copy. The complete document of 335 pages is provided in pdf format on CD.
- E-mail communication with Hanna Ng, Director of Preclinical Safety, SRI International